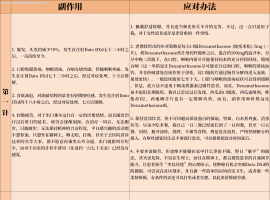
One of the main reasons of resistance to EGFR tyrosine kinase inhibitors (TKIs) is that there are alternative mechanisms for persistent activating EGFR downstream signaling, including both RAS/Erk and PI3K/Akt kinase pathways. Therefore, simultaneous inhibition of both pathways would be necessary to reduce tumor cell survival more effectively. One of the candidate combinations is concurrent use of EGFR-TKIs and statins, which are irreversible inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase and have been used to treat hypercholesterolemia through blocking the mevalonate biosynthesis pathway. Beside the cholesterol lowering effect, statins have been shown to induce apoptosis in several tumor types. It affects the synthesis of other products of the mevalonate pathway such as isoprenoids, which are used as substrates for prenylation. Attachment of isoprenoids to RAS proteins facilitates their anchoring to the cell membrane where they carried out their roles. By interrupting the biosynthesis of mevalonate, statins inhibit activation of RAS and downstream signaling cascades, including the RAF/MEK/ERK and PI3K/AKT, which play critical roles in regulation of cell survival and proliferation. Therefore, it seems to be a promising therapeutic approach overcoming tumor resistance to EGFR-TKIs, which is associated with RAS activation.
According to the recent clinical result of phase II trial, a randomized phase II study of gefitinib with or without simvastatin in previously treated patients with advanced NSCLC conducted by Han et al.37 gefitinib plus simvastatin combination produced higher response rates than gefitinib alone in patients with non-adenocarcinoma (5/13 [39%] v 1/13 [8%], P=0.06). This finding suggests that simvastatin may enhance sensitivity to gefitinib in non-adenocarcinoma that is relatively resistant to gefitinib. Moreover, by Mantha et al.35 demonstrated that the combination of gefitinib and lovastatin showed significant synergic cytotoxic effects in vitro in a total of 16 squamous cell carcinomas, NSCLC, and colon carcinoma cell lines. Of special interest, these cell lines did not possess the activating mutations of EGFR, which confer increased sensitivity to gefitinib. Nevertheless, combining lovastatin with gefitinib induced more significant inhibition of AKT activation than either agent alone. Additionally, lovastatin significantly enhanced the sensitivity to gefitinib treatment regardless PTEN loss in glioblastoma cell lines. These results suggest that statins can augment EGFR inhibition.
|
 需要加论坛患者群的朋友们看这里
本帖最后由 青菜567 于 2024-7-22 17:38 编辑
与癌共舞小助手-29新进论坛需要进入患
需要加论坛患者群的朋友们看这里
本帖最后由 青菜567 于 2024-7-22 17:38 编辑
与癌共舞小助手-29新进论坛需要进入患
 亲历“天价”肺癌药物临床试验,3个
作者:林晓清
2024年11月26日,身为肺腺癌Del19缺失,晚期患者的我,正式开始接受Dato
亲历“天价”肺癌药物临床试验,3个
作者:林晓清
2024年11月26日,身为肺腺癌Del19缺失,晚期患者的我,正式开始接受Dato
 奥希替尼耐药后,我加入新药临床试验
作者:林晓清
二零二四年十月二十一日,我按照和Professor.Paul De Souza的预约,第三
奥希替尼耐药后,我加入新药临床试验
作者:林晓清
二零二四年十月二十一日,我按照和Professor.Paul De Souza的预约,第三
 肺癌历经手术-复发-脑转移,如今母亲
讲述者:一只阿狸整理者:pear
熟悉我家的朋友都知道,我母亲虽然是IB期肺癌,但是很
肺癌历经手术-复发-脑转移,如今母亲
讲述者:一只阿狸整理者:pear
熟悉我家的朋友都知道,我母亲虽然是IB期肺癌,但是很
 3A术后一年,别的医院ct报告
请大家帮忙看看是不是有问题,目前还在免疫治疗
3A术后一年,别的医院ct报告
请大家帮忙看看是不是有问题,目前还在免疫治疗





 显身卡
显身卡





